Profile Fitting Search-Match (PFSM)
Go to Match! Features Overview...
Previous: Guided operations...
Next: Rietveld refinement...
"Profile fitting search-match" (PFSM) is a powerful, innovative
alternative to the proven peak-based search-match functionality for qualitative phase analysis.
PFSM fits the profile calculated from every candidate entry in the reference database to your experimental diffraction pattern on the fly. Afterwards, it creates a ranking list (candidate list) so that the entries/phases which fit best (i.e. have the highest FoM value) are located at the top. Finally, the user can evaluate the entries at the top of the list and select those which are most likely to be present in the sample ("matching entries/phases").
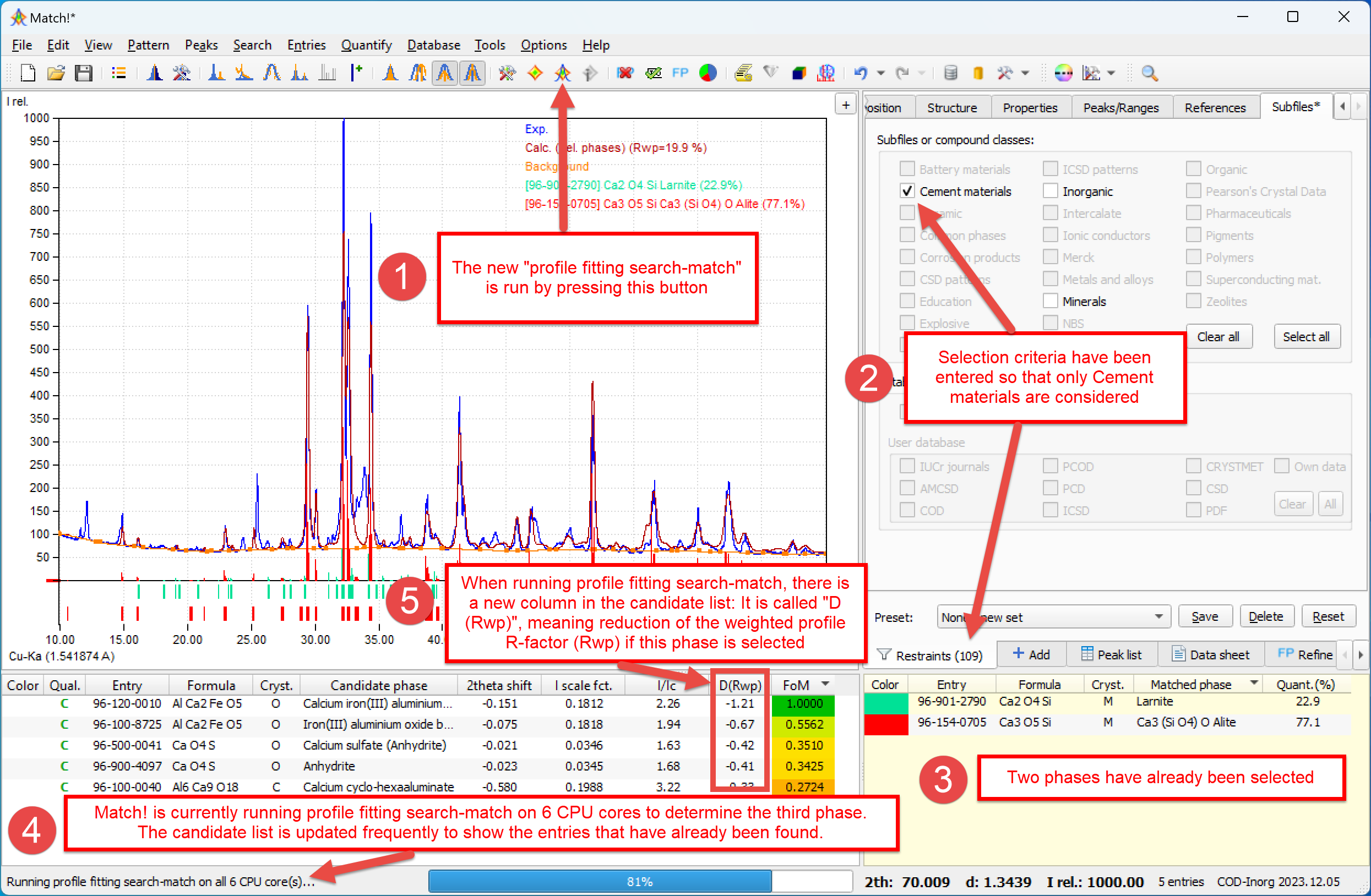
Here is a screenshot showing the profile fitting search-match function in action on a Cement sample:
You can also watch a video, showing the new Match! version 4 in action:
Advantages of PFSM
One of the main advantages of PFSM is that the sometimes ambiguous peak searching is no longer required for qualitative phase analysis, thus avoiding the ambiguities that may arise from this.
Due to this, the success rate of PFSM is definitely higher than the "classical" (peak-based) approach (especially with minor or trace phases), provided that the background is set correctly before the calculation.
Another important advantage (compared to the Rietveld refinement search-match approaches cited below) is that PFSM in Match! can use arbitrary peak data reference databases, like the COD, ICDD PDF-2, PDF-4, own diffraction patterns etc. The knowledge of atomic parameters for the candidate phases is not required.
How it works
PFSM in Match! works without additional software, i.e. no Rietveld software (like FullProf) is required. To be clear,
no Rietveld refinement is actually performed, because no structural parameters are refined.
Instead, Match! calculates a profile pattern for every entry of the reference database (or answer set),
fits both the intensity scale factor as well as the 2theta shift in order to get an optimum agreement with the experimental profile diffraction pattern, and finally stores the Rwp value reduction that could be achieved by doing so.
The background is not refined during the PFSM calculation, in order to avoid any ambiguities that may result when doing so. Hence, it is rather important to make sure that the orange background curve in the pattern graphics is in perfect agreement with the background that you estimate "by eye" before running the PFSM calculation.
Display of results
During the PFSM calculation, all candidate entries for which the profile fitting has already been performed are displayed in the candidate list (at least if the corresponding option is active).
The candidate entries are ranked according to decreasing reduction of the Rwp value (D(Rwp)) that could be achieved during the corresponding fit (taking into account the already selected phases).
Due to the fact that the results are ranked according to decreasing reduction of the Rwp value, the phases are normally identified in the same order as they contribute to the diffraction profile. Normally, the phase with the highest amount is selected first and the phase with the lowest amount last (also depending on their scattering power, of course).
Calculation times
While the success rate of PFSM is definitely higher than the "classical" (peak-based) approach (especially with minor or trace phases),
there is certainly one main drawback: The calculation time is significantly longer.
In order to compensate for this
to a certain degree, PFSM is by default run in parallel on all available cores of your CPU.
It is also possible to restrict the calculation to a lower number of CPU cores (or even to a single core), in order to keep your computer more "responsive" for other tasks.
Here are a few hints what you can do to speed-up the calculation:
-
Use any additional information available about the sample, e.g. by selecting elements that may be present or can be excluded (known e.g. from XRF), and/or by selecting a suitable application context (e.g. "Cement materials") as subfiles.
-
Calculations may also be sped-up by removing (trimming) the high-angle part of your diffraction pattern (e.g. the range beyond 70-80 degrees 2theta).
-
If available, use a computer with more CPU cores.
Please note though that the improvement that may be achieved by this can be lower than expected, especially if the individual profile-fitting calculations take only a very short time. The main reason is that in these cases the calculation time will be dominated by the internal administrative tasks to distribute the calculations to the individual cores and collecting the results afterwards.
-
Although we do not generally recommend it, you can also speed-up the PFSM calculations by reducing the resolution of your diffraction data. You can do so by running the "Decrease resolution..." command from the "Pattern" menu.
Some importants facts and hints
-
During PFSM, the intensity scale factor and 2theta-correction parameters of already selected (match list) entries are refined along with each individual candidate entry, but the resulting values (intensity scale factor and 2theta shift) are not retained in the match list entries.
-
Due to the inclusion of the already selected phases (match list entries) in the profile fitting calculation, PFSM takes longer when more entries are present in the match list.
-
The FoM values in the candidate list obtained from PFSM are relative, with the best-matching entry (the one that reduces the Rwp most) being attributed FoM=1.0.
-
As a consequence, there still may be candidate entries with a "promising" high FoM value even if all phases in the sample have been selected! In order to avoid selecting "false positives" in this situation, please also check the values (columns) "I scale fact." and "D(Rwp)" in the candidate list. If these values are rather low (close to zero), it is not very likely that these phases are present.
The background is that especially after all phases present in the sample have been selected, there may remain entries in the candidate list with low intensity scale factor but a lot of peaks that "mimic" the background and the noise in the diffraction data.
-
You can adjust both the minimum intensity scale factor as well as the minimum Rwp reduction required for entries being displayed in the candidate list in the Search-Match options.
-
Once a PFSM calculation has finished, the following options will be activated automatically:
-
Do not mismatch PFSM with profile fitting! PFSM uses the profile calculated from reference database entries, while profile fitting is operated on the experimental peak data.
References
Profile-fitting search-match as it is now implemented in Match! has not been invented "out of the blue" but has been inspired by other approaches, most of which use Rietveld refinement for qualitative phase analysis:
- Profex / BGMN using automatic Rietveld refinements for each candidate phase
- Luca Lutterotti: "Full Profile Search Match (FPSM)": Lutterotti, L. et al., "Full-profile search-match by the Rietveld method", J. Appl. Cryst. 52 (2019).
- SNAP-1D: C. J. Gilmore, G. Barr and J. Paisley, "High-throughput powder diffraction. I. A new approach to qualitative and quantitative powder diffraction pattern analysis using full pattern profiles", J. Appl. Cryst. 37, 231-242 (2004)
and
Barr, G., Gilmore, C. J. & Paisley, J., "SNAP-1D: a computer program for qualitative and quantitative powder diffraction pattern analysis using the full pattern profile", J. Appl. Cryst. 37, 665-668 (2004).
Go to Match! Features Overview...
Previous: Guided operations...
Next: Rietveld refinement...
|